

La diabetes mellitus neonatal (DN) se define como la hiperglucemia persistente, de inicio en los primeros 6 meses de vida. Se engloba dentro de las diabetes monogénicas aquellas causadas por una alteración de un sólo gen, que afecta al desarrollo y la función de las células beta del páncreas llevando a una alteración en la producción de insulina.
La mayoría de los pacientes con DN desarrollan la sintomatología en los 6 primeros meses de la vida, pero en algunos casos se puede presentar hasta los 12 meses. Su incidencia varía entre 1:90.000-160.000 recién nacidos, y es determinante realizar un diagnóstico genético de cada subtipo de DN ya que permitirá personalizar el tratamiento y condicionará el pronóstico.
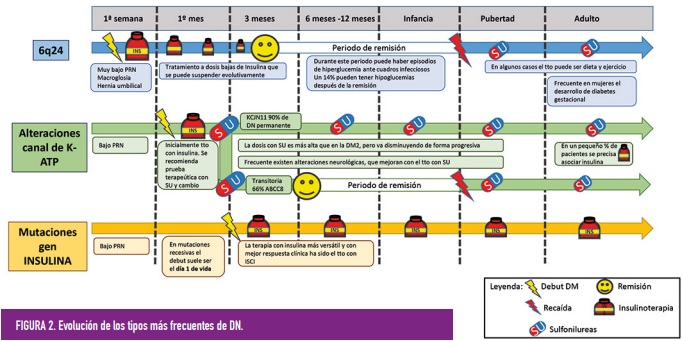
La evolución de la DN dependerá fundamentalmente del subtipo clínico diagnosticado.
Subtipos clínicos de diabetes neonatal monogénica
Existen dos subtipos clínicos: la forma permanente que requiere tratamiento continuado desde el diagnóstico y representan más del 50% de los casos, y la forma transitoria, que remite habitualmente en semanas o meses después de su aparición, pero que puede recidivar posteriormente a lo largo de la vida.
Las manifestaciones clínicas iniciales son poco específicas, incluyen un bajo peso al nacer debido a la disminución de la secreción de insulina durante el periodo intrauterino ya que la insulina ejerce un potente efecto promotor del crecimiento durante este periodo, deshidratación y una marcada hiperglucemia que puede o no asociar cetoacidosis diabética (CAD). En general, los anticuerpos contra los islotes pancreáticos son negativos (si son positivos hay que pensar en formas raras de DN monogénica como la secundaria a mutaciones en el factor de transcripción FOXP3) y la concentración de péptido C está disminuida. Pueden asociar alteraciones extra pancreáticas que pueden dirigir al diagnóstico de mutaciones en un determinado gen, aunque estas alteraciones pueden no estar presentes al inicio.
En este artículo vamos a centrarnos en las causas más frecuentes de DN y en su evolución clínica. Abordaremos la DN secundaria a anomalías en el cromosoma 6 (locus 6q24), la debida a alteraciones en los genes que codifican el canal de potasio voltaje dependiente y las secundarias a mutaciones en el gen de la insulina (gen INS).
Diabetes neonatal debida a anomalías en el cromosoma 6 (locus 6q24)
Se trata de la forma más frecuente de DN transitoria. El diagnóstico suele ser más precoz que en otros tipos de DN, frecuentemente durante la primera semana de vida. Suele asociar un peso muy bajo al nacer, generalmente por debajo del percentil 1. En un tercio de los casos asocian macroglosia y menos frecuentemente hernia umbilical. La remisión ocurre habitualmente casi en el 100% de los casos. El tratamiento suele ser con insulina, normalmente a dosis bajas, que se irá disminuyendo de forma progresiva, en general de forma rápida; la mayoría de los niños no requieren insulina a una edad media de 12 a 14 semanas. Como en la mayoría de estos pacientes tienen algún grado de función de la célula beta, no siempre es necesaria la terapia insulínica y pueden responder al tratamiento oral con sulfonilureas. Es frecuente que durante la infancia y ante episodios intercurrentes infecciosos se objetive hiperglucemia intermitente, sin embargo, hasta un 14% de los pacientes con anomalías en el cromosoma 6, pueden tener también hipoglucemias tras la remisión inicial. Tras la recidiva que ocurre en al menos 50 a 60% de los casos (en torno a los 14 años de edad, aunque puede variar desde la infancia temprana, a la pubertad o incluso, más adelante) son efectivos tratamientos orales como las sulfonilureas. Las mujeres con alteraciones del locus 6q24 tienen mayor riesgo de desarrollar diabetes gestacional. Hay que mentalizar a los padres del riesgo de recidiva. Estos pacientes se benefician de la determinación anual de la HbA1c.
Diabetes neonatal debida a mutaciones en los genes del canal de potasio (KATP)
La segunda causa más frecuente de DN transitoria y primera causa de DN permanente son las producidas por las anomalías en dos genes que codifican el canal de potasio ATP. Estos canales están situados en las membranas de algunas células incluyendo la célula ß. KCNJ11 codifica la subunidad Kir6.2 y ABCC8 codifica la subunidad SUR1. Alteraciones en cualquiera de estos genes producen una apertura inadecuada del canal de potasio que impide la despolarización de la membrana celular y la liberación de la insulina (figura 1)
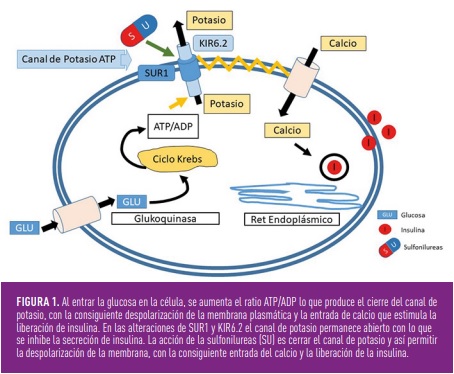
Los pacientes con alteraciones en el canal de potasio-ATP son diagnosticados, generalmente, durante los 3 primeros meses de la vida (edad media 9,6 semanas), y pueden asociar patología neurológica como dificultades de aprendizaje, trastorno de déficit de atención e hiperactividad, dificultades emocionales o trastornos de conducta. En casos de mutaciones graves en KCJ11 se puede desarrollar retraso mental y epilepsia (síndrome DEND). Tienen habitualmente una disminución moderada del peso neonatal, generalmente en los percentiles bajos de la normalidad. La diabetes puede ser permanente o transitoria. Los pacientes con mutaciones en Kir6.2 tienen DN permanente en el 90% de los casos. Mientras que en las mutaciones en el ABCC8 en el 66% son formas transitorias. Cuando la diabetes es transitoria, la remisión se produce pasados 3-6 meses, en algunos casos más tardíos (hasta los 18 meses de edad). En las formas transitorias se han descrito recidivas de la diabetes en etapas posteriores de la vida. (figura 2)
La DN causada por alteraciones del canal de potasio-ATP es un magnífico ejemplo de la medicina personalizada ya que el diagnóstico genético ha permitido el cambio de tratamiento de insulina a sulfonilureas (SU). Las SU actúan sobre el canal de potasio promoviendo su cierre, lo que permite la liberación de insulina por parte de la célula. La glibenclamida ha sido el tipo de SU más frecuentemente utilizada pero también se han probado con buenos resultados glipizida, glicacida, tolbutamina o glimepiride. El uso de SU incrementa la liberación de insulina por lo que aumenta el riesgo de hipoglucemia, especialmente si el niño disminuye la ingesta. Sin embargo, el riesgo de hipoglucemia es más bajo que en aquellos individuos que son tratados con insulina. La dosis requerida de SU por Kg. de peso es superior a la utilizada en la diabetes tipo 2 del adulto. El gen ABCC8 que codifica la proteína SUR11 es crucial para la función retiniana y la glibenclamida confiere una acción directa de nuero protección a través de mecanismos mediados por el SUR-1.
En un estudio publicado por Bowman en 2018, en una cohorte de 81 pacientes con al menos 10 años de seguimiento clínico, tratados con SU, se objetiva una mejoría significativa en el control clínico, disminuyendo la hemoglobina glicosilada de 8,1 a 5,9% tras un año de tratamiento, y 6,4% en el último control clínico. No se registraron episodios de hipoglucemia grave. Sí que se notificaron otros efectos secundarios como diarrea, náuseas, pérdida de apetito, esteatosis hepática o decoloración dental. El 9% de los pacientes tenía complicaciones microvasculares: retinopatía, microalbuminuria, proteinuria y neuropatía. En estos pacientes se observó que el cambio de terapia se había producido más tardíamente (20,5 años vs 4,5 años).
Diabetes neonatal debida a mutaciones en el gen de la insulina
La segunda causa más frecuente de DN permanente son las mutaciones en el gen de la insulina (gen INS) siendo las causantes del 20% de las DN permanentes. La edad media al diagnóstico son las 10 semanas de edad y el 30% de los pacientes debutan en CAD. Los niveles de péptido C están disminuidos o son indetectables y el peso al nacimiento se sitúa en los límites bajos de la normalidad. Si se trata de una mutación recesiva del gen INS, el bajo peso al nacimiento es mucho más acusado, el debut de la diabetes mucho más temprano, frecuentemente en el primer día de vida, y hasta un 26% de los casos son transitorios. No asocian otras patologías extra pancreáticas. Aunque la mayoría de las mutaciones de la insulina son de novo (esporádicas) un 20% tiene historia familiar de diabetes monogénica autosómica dominante.
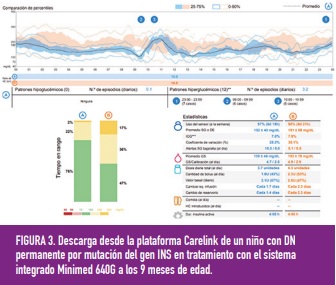
El tratamiento generalmente es crónico con insulina. La terapia insulínica puede ser con MDI (múltiples dosis de insulina) o ISCI (infusión subcutánea continua de insulina/bombas de insulina). El tratamiento con ISCI tiene la ventaja de poder administrar dosis de insulina más pequeñas y precisas, así como correcciones mínimas en caso de hiperglucemia. Los sistemas integrados en los que se asocian la monitorización continua de glucosa con la ISCI y un algoritmo añaden la ventaja de la parada en predicción de hipoglucemia, lo que es fundamental en estos niños tan pequeños y generalmente con necesidades de insulina bajas. En ocasiones se requiere utilizar diluciones de la insulina.
Rabbone y cols. en el 2016, revisaron las publicaciones de casos clínicos de pacientes con DN monogénica tratados con insulina y constataron que la dosis media utilizada en ISCI variaba entre 0,2 y 1,4 u/kg/día, y que la tasa basal oscilaba entre el 20 y el 76% de la dosis total de insulina del día. Tras dicha revisión concluyeron que el tratamiento con ISCI en la DN era efectivo y seguro. En ese mismo año, Ortolani y cols. publicaron la evolución clínica durante los 2 primeros años de vida en tres pacientes con DN por mutaciones del gen INS en tratamiento con sistemas integrados, concluyendo que también eran efectivos y seguros y que mejoraban las prestaciones de la ISCI aislada (figura 3).
Otros tipos de diabetes neonatal
Se han descrito más de 30 subtipos de DN monogénica. Existen DN asociadas a síndromes más complejos en los que se asocian otras alteraciones. Si hay consanguinidad, las formas más frecuentes son el síndrome de Wolcott-Rallison que asocia un comienzo precoz de diabetes mellitus con alteraciones hepáticas y renales y displasia esquelética y las mutaciones homocigotas del gen de la glucoquinasa (GCK). Entre las formas más raras de diabetes neonatal permanente se han encontrado mutaciones en otros genes como GATA6, GLIS3 o PAX6. Hasta en un 20% de las diabetes neonatales permanente, hoy en día, no se encuentra la causa genética.
Conclusiones
- La evolución de la diabetes neonatal depende de la alteración genética subyacente (figura 2).
- Se debe realizar, sin demora, el estudio genético en todos los casos de debut diabético en niños menores de 6 meses y también se debe considerar el estudio genético en los niños diagnosticados de diabetes entre los 6 y los 12 meses de vida, especialmente en aquellos que tengan autoinmunidad pancreática negativa u otros hechos sugestivos de diabetes monogénica.
- El diagnóstico genético de la DN da una información esencial sobre las opciones de tratamiento, alteraciones asociadas y evolución de la diabetes que puede tener un significado clínico relevante.
- En los casos de alteraciones del canal de potasio se debe realizar el cambio de tratamiento de insulinoterapia a sulfonilureas (especialmente glibenclamida). Esta mejora las anomalías neurosicológicas de los niños con DN debidas a mutaciones del KCNJ11 o ABCC8.
- La forma más fisiológica de aportar insulina en el periodo neonatal son los sistemas de administración subcutánea continua de insulina.
Referencias
- Lemelman MB, Letourneau L, Greeley SAW. Neonatal diabetes mellitus: an update on diagnosis and management. Clin Perinatol. 2018;45(1):41–59.
- Dahl A, Kumar S. Recent Advances in Neonatal Diabetes. Diabetes Metab Syndr Obes. 2020;13:355-364.
- Greeley SAW, Polak M, Njolstad PR et al. ISPAD Clinical Practice Consensus Guidelines 2022: the diagnosis and management of monogenic diabetes in children and adolescents. Pediatr Diabetes. 2022;23:1188-1211. DOI: 10.1111/pedi.13426.
- Docherty L, Kabwana S, Lehman A, Hawke E, Harrison L, Flanagan S et al Clinical presentation of 6q24 transient neonatal diabetes mellitus (6q24 TNDM) and genotype–phenotype correlation in an international cohort of patients. Diabetologia. 2013;56:758–762
- Bonfanti R, Iafusco D, Rabbone I, et al. Differences between transient neonatal diabetes mellitus subtypes can guide diagnosis and therapy. Eur J Endocrinol. 2021;184(4):575-585. doi:10.1530/EJE- 20-1030
- Flanagan SE, Mackay DJ, Greeley SA, et al. Hypoglycaemia following diabetes remission in patients with 6q24 methylation defects: expanding the clinical phenotype. Diabetologia. 2013;56(1):218–221.
- Rabbone I, Barbetti F, Marigliano M, et al. Successful treatment of young infants presenting neonatal diabetes mellitus with continuous subcutaneous insulin infusion before genetic diagnosis. Acta Diabetol. 2016;53(4):559–565. doi: 10.1007/s00592-015-0828-7
- Rabbone I, Barbetti F, Gentilella R, et al. Insulin therapy in neonatal diabetes mellitus: a review of the literature. Diabetes Res Clin Pract. 2017;129:126–135
- Ortolani F., Piccinno E., Grasso V., Papadia F., Panzeca R., Cortese C., et. al.: Diabetes associated with dominant insulin gene mutations: outcome of 24-month, sensor-augmented insulin pump treatment. Acta Diabetol 2016; 53:499-501.
- Bowman P, Sulen A, Barbetti F, et al. Effectiveness and safety of long-term treatment with sulfonylureas in patients with neonatal diabetes due to KCNJ11 mutations: an international cohort study. Lancet Diabetes Endocrinol. 2018;6(8):637–646. doi: 10.1016/S2213-8587(18)30106-2



